
В этом тексте мы затронем то, как ученые научились производить антитела для лекарственного применения, а также модифицировать их. Поскольку антитело — очень сложная молекула, обладающая пространственной структурой, определяющей ее функцию, — антитело нельзя синтезировать химически, а нужно обязательно использовать для этого биологические системы, например, клеточные культуры животных, в которые заложены все необходимые средства для производства таких сложных белков. В этой статье мы рассмотрим современные подходы в биоинженерии антител. Мы расскажем о том, как биотехнологи создают антитела к нужным мишеням: о генной инженерии антител, создании их «с нуля», оптимизации структуры и производстве в клеточных культурах животных (рис. 1).
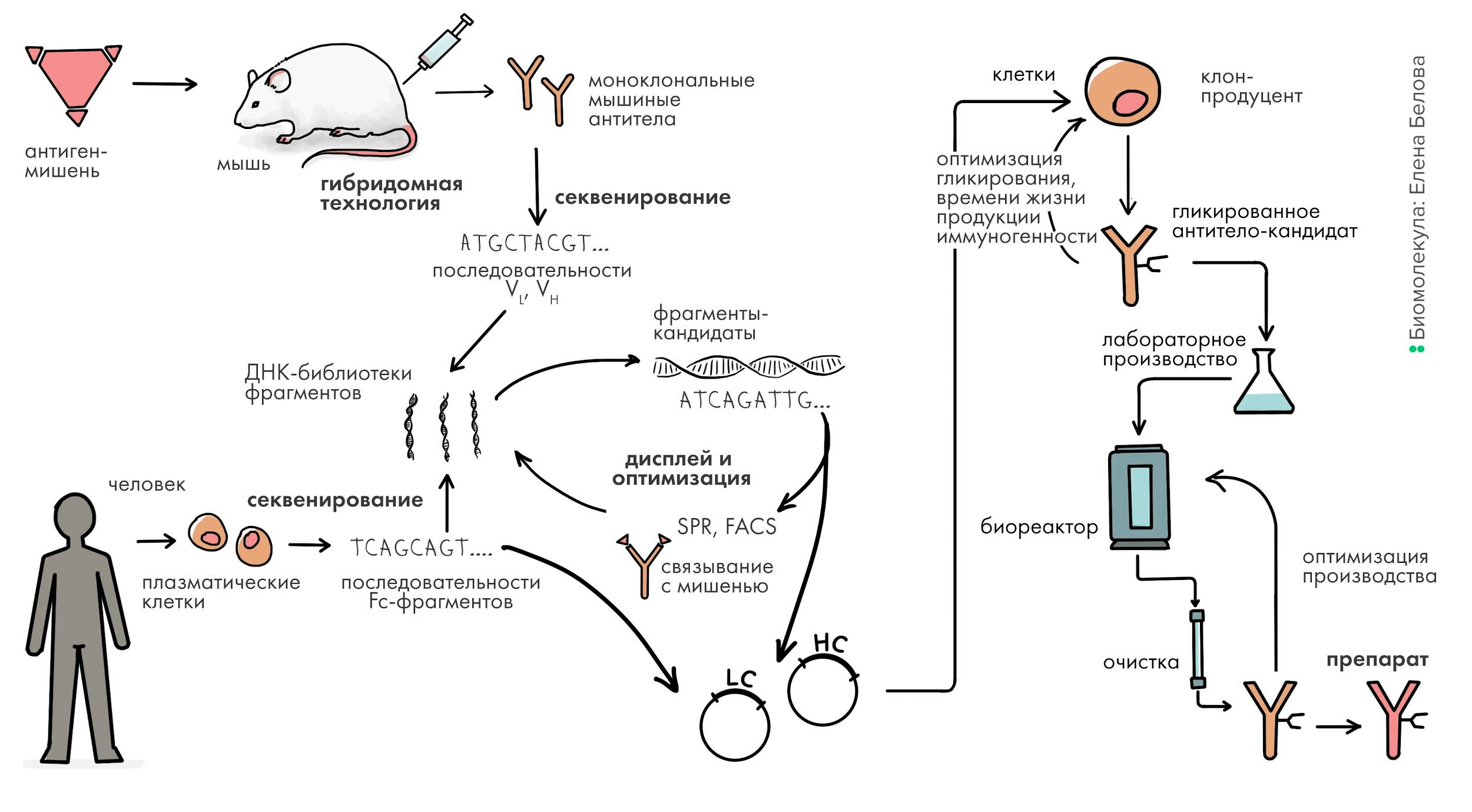
Рисунок 1. Общая схема разработки антитела
Для того чтобы клетка производила антитело, необходимо ввести в нее гены, кодирующие его тяжелые и легкие цепи. Также нужно, чтобы генетическая конструкция, которая вводится в клетку (ее называют вектором), содержала ряд вспомогательных элементов, обеспечивающих наиболее эффективную транскрипцию и трансляцию гена в клетке (рис. 2). Уже на данном этапе необходимо продумать весь путь разработки антитела вплоть до выхода на рынок. Для этого еще на стадии конструирования антитела создают Target product profile — документ, который со временем превратится в инструкцию к препарату. Именно он определяет, каким пациентам будет показано антитело, с какой частотой оно будет вводиться, а значит, каков должен быть его формат и другие параметры, о которых мы расскажем ниже.
Путем изменения генетических конструкций, кодирующих антитело, можно влиять на такие его характеристики, как способ действия (будет оно убивать клетку-мишень или только препятствовать проведению сигнала), селективность к мишени, время выведения из организма и др. Если необходимо модифицировать специфичность или селективность антитела, то изменению подлежат вариабельные фрагменты антител, отвечающие за связывание с антигеном (Fab-фрагменты). Если же нужно изменить другие параметры — время полужизни антитела, его механизм действия, — модифицируют константные участки (Fc-фрагмент).
Генная инженерия антител
Обычно создание генно-инженерной конструкции для экспрессии антитела начинается с выбора системы экспрессии, вектора и оптимизации нуклеотидной последовательности, кодирующей антитело. Необходимо откорректировать состав кодонов таким образом, чтобы экспрессия антитела была максимальной. Аминокислотный состав белка оптимизируют для снижения вероятности выпадения в осадок, агрегации, а также иммуногенности, о чем мы подробнее расскажем далее. Кроме того, важно обеспечить правильное сворачивание (фолдирование, упаковку) цепей и их соединение между собой. Сейчас существуют программы, позволяющие оптимизировать нуклеотидную последовательность генно-инженерных конструкций для продукции антител in silico, хотя, конечно, последнее слово остается за экспериментальными данными.
В качестве вектора используют плазмиды — кольцевые ДНК, кодирующие как сам ген белка, так и вспомогательные элементы. Так, для экспрессии в эукариотических клетках используют промотор цитомегаловируса (CMV), обеспечивающий высокую эффективность транскрипции.
Поскольку антитело состоит из двух полипептидных цепей — легкой и тяжелой, — используют либо двухплазмидную систему экспрессии, либо один вектор, содержащий оба гена. Последний вариант предпочтительнее, так как позволяет точнее контролировать соотношение продукции легких и тяжелых цепей. Было показано, что в природных условиях в B-клетках легкие цепи синтезируются в бóльшем количестве, чем тяжелые, и такое соотношение скоростей синтеза наиболее благоприятно для продукции антител.
Вектор должен также содержать ген устойчивости к антибиотику, в присутствии которого выращивают клетки. Это нужно, чтобы клетки не избавлялись от вектора — при добавлении антибиотика клетки, не несущие вектор, погибнут. Хотя антитело синтезируется в клетках млекопитающих, предварительные манипуляции с вектором (например, вставка туда гена и клонирование, то есть получение одинаковых копий вектора) проводят в бактериях . На рис. 2 показан вектор с двумя генами устойчивости к антибиотикам — один для бактерий, второй для эукариотических клеток.
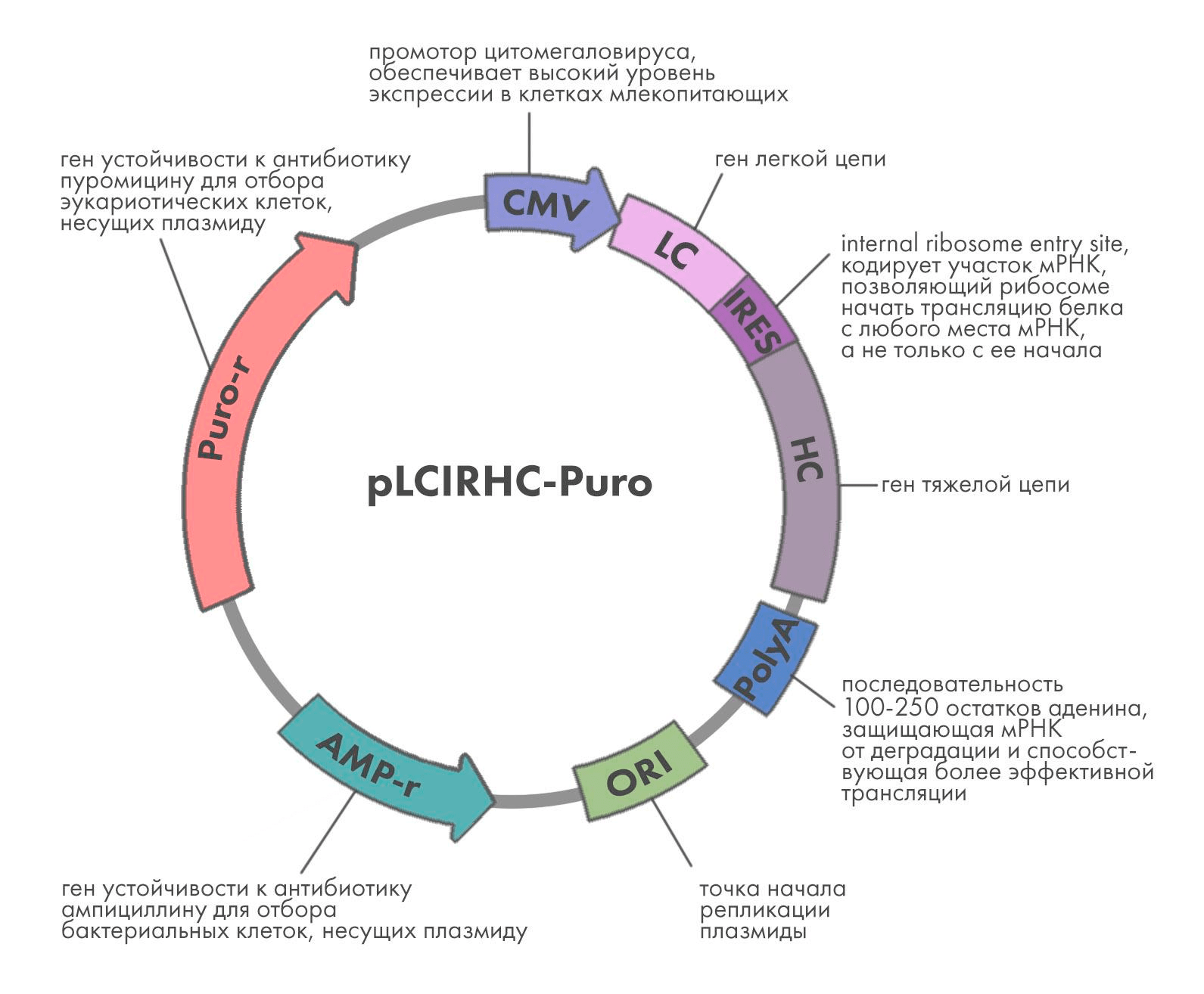
Рисунок 2. Схема плазмиды для экспрессии антител в клетках млекопитающих
Получение вариабельных фрагментов антител
Перейдем к тому, как получаются последовательности вариабельных фрагментов антител, которые и определяют их специфичность и селективность.
Исследователи пока не могут предсказать, какая именно последовательность вариабельного фрагмента антитела будет оптимальной для связывания антигена. Антиген, как правило, представляет собой белковую молекулу, и площадь поверхности контакта антитела с антигеном слишком велика для моделирования in silico (рис. 3), которое также осложняется наличием молекул воды. Поэтому приходится использовать биологические объекты, у которых есть способность очень тонко настраивать последовательность аминокислот антитела для обеспечения высочайшей аффинности к антигену.
Биологические процессы настолько сложны, что порой их намного удобнее смоделировать на компьютере, нежели пытаться повторить в пробирке.
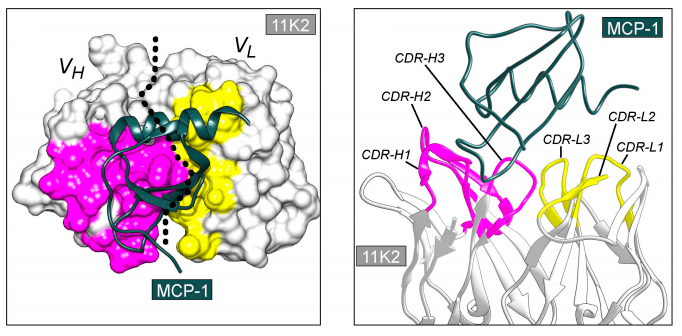
Рисунок 3. Кристаллическая структура комплекса антитела 11К2 (серый, розовый и желтый цвета) и антигена MCP-1 (зеленый цвет) в двух проекциях. Структура иллюстрирует сложность поверхности контакта антитело—антиген.
Методы получения антител подразделяются на гибридомные и дисплейные. Гибридомный метод, использует для получения антител организм млекопитающего, который после иммунизации (введения антигена) вырабатывает антитела с высокой аффинностью и селективностью. В дальнейшем эти антитела или их гены можно выделить, отсеквенировать и модифицировать с целью придания им нужных фармакологических свойств.
Дисплейные методы
Дисплейные методы используют в качестве исходного материала широкий репертуар генов, кодирующих антитела. Самым распространенным методом является фаговый дисплей. Метод представления (отсюда название — «дисплейный») пептида на поверхности бактериофага М13 был изобретен в 1985 году. В 1989 г. научились клонировать гены иммуноглобулинов, что позволило обойтись без гибридомной технологии для получения антител. Первоначально в бактериях синтезировали Fab или scFv фрагменты антител, создавая библиотеки клонов, и проводили вручную отбор наиболее аффинных клонов с помощью радиоактивно-меченного антигена. Однако это была очень трудоемкая и неэффективная процедура. В 1990 году впервые использовали дисплейную технологию представления фрагментов антител на поверхности фагов, а в 1991 создали первую библиотеку вариабельных фрагментов. Первым антителом, созданным с помощью технологии фагового дисплея, стала «Хумира» (адалимумаб компании Abbott) против мишени TNF для лечения ревматоидного артрита и болезни Крона. «Хумира» зарегистрирована в 2002 году, и уже долгое время возглавляет список лекарств — лидеров продаж.
Получение антитела дисплейным методом in vitro схематически подражает эволюционному процессу формирования антител в организме:
- Сперва необходимо породить разнообразие генов, кодирующих вариабельные фрагменты — так получаются библиотеки антител.
- Затем нужно экспрессировать эти гены, то есть преобразовать генотип в фенотип.
- Потом нужно осуществить селективное давление, которое бы заставило генотип эволюционировать.
- И наконец необходимо амплифицировать полученный результат.
Таким образом, в процессе присутствуют все составляющие эволюции — наследственность, изменчивость и отбор. Рассмотрим эти шаги подробнее.
Качество получаемой библиотеки фрагментов зависит от способности получить широкий репертуар разнообразных и при этом качественных антител. Существует два принципиально разных подхода к получению библиотек: естественный (наивный) и синтетический.
Естественные библиотеки вариабельных доменов антител получают методом полимеразной цепной реакции природных генов с обратной транскрипцией (RT-PCR) из лимфоидных тканей или периферической крови людей, а также других животных. Преимущество этого метода в том, что полученные антитела будут в правильной конформации, так как их гены кодируют функциональные антитела. Однако недостаток в том, что разнообразие последовательностей ограничено охватом естественной иммунной системы, в которой существует определенная неравномерность использования тех или иных последовательностей. Также фрагменты из природных библиотек сильно отличаются по качеству и непредсказуемы по составу; многие из них могут оказаться недостаточно стабильными или неподходящими по другим причинам. Репертуар естественных библиотек составляет 107–1011 фрагментов.
Синтетические библиотеки создают путем встраивания искусственно синтезированной ДНК в последовательности, кодирующие вариабельные домены. ДНК синтезируется таким образом, чтобы вносить совершенно случайные (рандомизированные) мутации в получающиеся фрагменты антител и тем самым расширять разнообразие определяющего комплементарность сайта (complementarity-determining region, CDR). Синтетические библиотеки позволяют использовать определенную коровую (germline) структуру вариабельного домена, про которую известно, что она наиболее представлена в каждом индивидууме, неиммуногенна и стабильна. Однако введение полностью синтетических участков CDR может привести к неправильному сворачиванию и агрегации белка. Потребовалось время для отработки подходов к определению того, какие CDR лучше использовать. Репертуар синтетических библиотек, как правило, доходит до 109–1011 фрагментов.
Далее начинается самое интересное: как дисплейная технология позволяет связать генотип с фенотипом и отобрать клоны с нужными свойствами. Гены всех фрагментов антител вставляют в геном бактериофага в одно и то же положение — внутрь гена, кодирующего белок оболочки фага. Потом гены переносят в кишечную палочку Escherichia coli (это называется трансдукцией) и заражают вспомогательным фагом, который вызывает сборку бактериофага, содержащего на поверхности фрагмент антитела. В результате получается библиотека бактериофагов, у каждого из которых на поверхности экспрессируется фрагмент антитела, соответствующий гену, который попал в данного бактериофага. Препарат библиотеки добавляют к иммобилизованному на поверхности пластиковой пробирки антигену, против которого надо создать антитела, и проводят отмывку. На поверхности остаются только бактериофаги, содержащие вставку гена фрагмента антитела с высоким сродством к антигену. Чтобы отобрать самые высокоаффинные клоны, связавшиеся с антигеном, фаги снимают с поверхности и снова заражают ими E. coli (рис. 4). После нескольких этапов отбора ДНК полученных фагов секвенируют и узнают последовательность, кодирующую самые аффинные фрагменты.

Рисунок 4. Процесс селекции нужных генотипов в соответствии с аффинностью к антигену. а — Бактериофаг, обеспечивающий связь генотип—фенотип. Ген фрагмента антитела (розовый) вставляется в фаговую ДНК, что приводит к экспрессии на поверхности фага белка — фрагмента антитела. б — Схема фагового дисплея. 1. Фаговую библиотеку, состоящую из 106–1011 клонов, инкубируют с иммобилизованным антигеном. 2. Не связавшиеся фаги удаляют промывкой. 3. Связавшиеся фаги снимают с поверхности. 4. E. coli заражают фагом для амплификации полученных кандидатов. 5. Клетки высевают на среду с антибиотиком и амплифицируют. Процесс повторяют 2–3 раза для обогащения популяции антительными фрагментами, специфичными к антигену.
Важное преимущество использования дисплейных методов по сравнению с гибридомными — возможность отбирать только те антитела, которые НЕ связываются с ненужными белками, например, с близкими к мишени, но которые не надо блокировать, чтобы не вызывать токсичность. Для этого достаточно иммобилизовать теперь уже нежелательный белок и отбирать те фаги, которые не связались с ним.
Также бывает полезно создать антитело, которое бы хорошо связывалось и с мышиным, и с обезьяньим, и с человеческим вариантами мишени — это обеспечивает быстрое успешное прохождение доклинических исследований и контроль безопасности до выведения потенциального лекарственного кандидата в клинические испытания.
Существуют и другие типы дисплеев, помимо фагового, например, рибосомный или дрожжевой. Здесь я не буду подробно на них останавливаться, так как принцип у них тот же: они обеспечивают связь гена, кодирующего фрагмент антитела, с его аффинностью к мишени, то есть с фенотипом, и позволяют производить отбор по заданным свойствам из огромного количества вариантов в библиотеке.
Созревание аффинности антител
После получения фрагментов-кандидатов разработчики приступают к более сфокусированной настройке аффинности, которая носит название созревание аффинности. В иммунной системе созревание аффинности происходит путем последовательного внесения мутаций в участки, кодирующие CDR, и селективного давления отбора в сторону бóльшей аффинности. In vitro процесс примерно такой же. Разработчики берут ген лидерного кандидата, вносят мутации в положениях соответствующих CDR и проводят скрининг на аффинность. Индивидуальные мутации, усиливающие аффинность, затем объединяют в одном клоне. Рибосомный дисплей позволяет довольно быстро проскринировать миллионы клонов, а использование нескольких раундов отбора имитирует естественный процесс созревания аффинности. В результате получаются антитела с аффинностью даже выше, чем у природных.
Полученная генетическая последовательность фрагмента антитела, содержащая вариабельные домены, объединяется с последовательностью, кодирующей константные участки тяжелой и легкой цепей (Fc-фрагмент). Если оптимизация вариабельного фрагмента касается аффинности и селективности антитела, то Fc-фрагмент отвечает за фармакокинетику и эффекторные функции антитела, о чем я более подробно расскажу далее.
Так выглядит простейшая схема получения антитела, однако на практике разработчики прибегают к модификации и совмещению различных подходов. Расскажу для примера, как была получена молекула уже упомянутой «Хумиры»: за основу взяли тяжелую цепь мышиного антитела против TNF, ее ген скомбинировали с библиотекой человеческих легких цепей и провели селекцию на связывание с человеческим TNF (hTNF). Полученное антитело-кандидат имело аффинность около 15 нМ. Далее провели созревание аффинности, которое позволило добиться аффинности на уровне 300 пМ.
Эпитопная специфичность
Если оптимизация аффинности — относительно простая задача, то инженерия эпитопной специфичности может оказаться достаточно трудным делом. От того, с каким участком мишени свяжется антитело, часто зависит итоговый эффект. Особенно это существенно в случае активирующих антител. Однако и для блокирующих антител это тоже важно: например, антитело с тщательно подобранной эпитопной специфичностью может предотвратить связывание мишени с одним из лигандов, не влияя на связывание с другими.
Один из самых ярких примеров важности определенной эпитопной специфичности — антитела против рецептора HER2, который часто повышенно экспрессируется в клетках рака молочной железы и выставляется на их поверхности. Антитело «Герцептин» (трастузумаб компании Roche/Genentech), одобренное в качестве первой линии терапии метастатического рака молочной железы с повышенной экспрессией HER2, связывается с С-концевым доменом HER2, который отвечает за связывание с другим рецептором, HER3, и препятствует активации последнего. Трастузумаб работает только у пациенток с высокой экспрессией HER2, и у большинства впоследствии развивается резистентность к препарату, то есть опухоль снова начинает расти. Компания Roche разработала антитело «Перджета» (пертузумаб), которое связывается с другим эпитопом HER2, отвечающим за его связывание с рядом рецепторов. Пертузумаб предотвращает активацию самого HER2, тем самым позволяя преодолеть резистентность к трастузумабу.
Один из методов создания антител к выбранному эпитопу состоит из двух этапов. На первом этапе проводится скрининг исходной библиотеки антительных фрагментов на связывание с выбранной мишенью, как описано выше. После того, как получены высокоаффинные фрагменты, проводится их скрининг с мутантной мишенью, у которой аминокислотные остатки в области эпитопа заменены на другие, препятствующие связыванию с ним. В ходе скрининга отбираются те антитела, которые НЕ связываются с мутантной мишенью. Это и будут антитела, специфичные к нужному эпитопу.
Выбор системы экспрессии
Выше было сказано, что систему экспрессии, то есть тип клеток, в котором будет происходить производство антитела, выбирают на раннем этапе, и ее выбор влияет на то, как должна выглядеть генетическая конструкция и как будут происходить дальнейшее масштабирование и оптимизация экспрессии. Наиболее распространенный во всем мире промышленный вариант системы экспрессии — клетки яичника китайского хомячка CHO (chinese hamster ovary). Клетки СНО дают хорошие выходы белка (как правило, 1–2 г с литра культуры, иногда до 10 г/л), обеспечивают правильное сворачивание и гликозилирование антител, способны к суспензионному росту (то есть в объеме, а не на поверхности) и хорошо адаптируются к изменению условий культивирования, что позволяет масштабировать процесс в различных объемах и использовать их в биореакторах. Другие клеточные линии включают NS0, Sp2/0 (обе — мышиной миеломы), HEK293 (клетки почки человеческого эмбриона) и PER.C6 (получены из человеческих фетальных ретинобластов). Также предпринимаются попытки производства антител в трансгенных растениях и животных, но они пока не дошли до коммерческого использования.
Оптимизация вариабельной части антитела — гуманизация
В случае классической гибридомной технологии антиген вводится, как правило, мыши, и в ходе естественного иммунного ответа вырабатываются антитела нужной специфичности. Помимо того, что естественная иммунизация не позволяет контролировать аффинность и селективность антитела, потому что мы не можем проконтролировать представление антигена в иммунной системе и последующий процесс продукции антител, есть еще более важная проблема: при гибридомной технологии мышиное антитело получается чужеродным по отношению к человеку из-за наличия других аминокислотных остатков по сравнению с антителами человека в определенных позициях константной части. Поэтому первое же коммерческое антитело против CD3 — OKT3 — вызывало у многих пациентов антительный ответ.
Такие антитела получили название HAMA — human anti-mouse antibodies, то есть «человеческие противомышиные антитела». Антительный ответ на введенное лекарство сильно снижает его эффективность, потому что приводит к выведению лекарства из организма и делает бессмысленным повторное введение препарата, так как во второй раз антительный ответ гораздо сильнее, и никакой пользы от лекарства точно не будет. Кроме того, антительный ответ может привести к серьезным аллергическим реакциям. Еще одна проблема не полностью человеческих антител — слабое взаимодействие мышиных антител с рецепторами человеческих клеток, которые обеспечивают некоторые из механизмов действия антител и длительность их пребывания в организме. Поэтому перед исследователями встала задача поиска технологий гуманизации антител, то есть приближения их к естественным человеческим аналогам.
Химерные антитела
Первым типом антител, приближенных к человеческим, стали химерные (рис. 5). В 1997 году был одобрен ритуксимаб — антитело против белка CD20 на поверхности В-лимфоцитов, предназначенное для лечения В-клеточных лимфом. Для его генерации полученные гибридомным методом вариабельные фрагменты объединили с человеческими константными доменами и таким образом достигли содержания человеческих участков более 70%. Включение ритуксимаба в схемы лечения лимфом произвело революцию, позволив не только увеличить время до прогрессирования заболевания, но и повысить общую выживаемость.
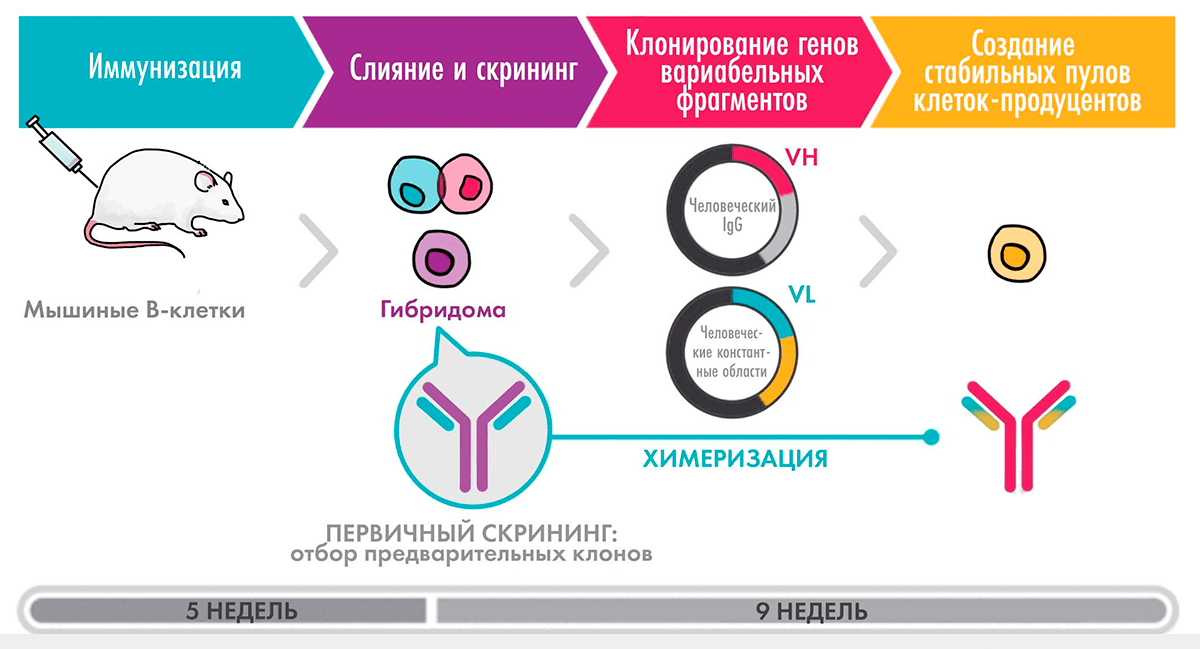
Рисунок 5. Схема получения химерного антитела
Рассмотрим более подробно процесс получения химерного антитела на примере инфликсимаба — препарата против TNF, который, как и рассмотренный выше адалимумаб, применяется для лечения ревматоидного артрита и болезни Крона. Вначале мышь иммунизировали человеческим TNF (hTNF) в сочетании с адъювантом — веществом, усиливающим иммунный ответ, — выделяли спленоциты и сливали их с клетками миеломы. Для отбора клонов клеток, экспрессирующих нужные антитела, использовали радиоиммунный метод с иммобилизованным hTNF. Затем после построения геномных библиотек с использованием фаговых векторов и скрининга путем саузерн-блоттинга выделяли гены, кодирующие легкие и тяжелые цепи антитела. После этого получали плазмиду, содержащую гены мышиных вариабельных участков, объединяли с генами человеческих константных областей (полученных ранее секвенированием генов В-клеток человека) и вводили ее в клетки мышиной миеломы для продукции химерного антитела.
Гуманизированные антитела
Хотя химерные антитела оказались более эффективными и менее иммуногенными, чем полностью мышиные, они все же вызывают HACA-ответ — образование human anti-chimeric antibody, то есть человеческих антител против химер. Поэтому с 1980-х годов начали разработку гуманизированных антител, у которых чужеродны человеку только CDR и отдельные позиции FR-регионов вариабельных доменов, а остальные части, включая каркас, почти полностью человеческие (рис. 6). Первым таким антителом в разработке стал алемтузумаб против антигена лимфоцитов CD52. Это лекарство применяется для лечения некоторых гемобластозов и рассеянного склероза. Оно было получено на основе крысиного антитела, чьи CDR перенесли в человеческие домены VH и VL уже существующего человеческого антитела.
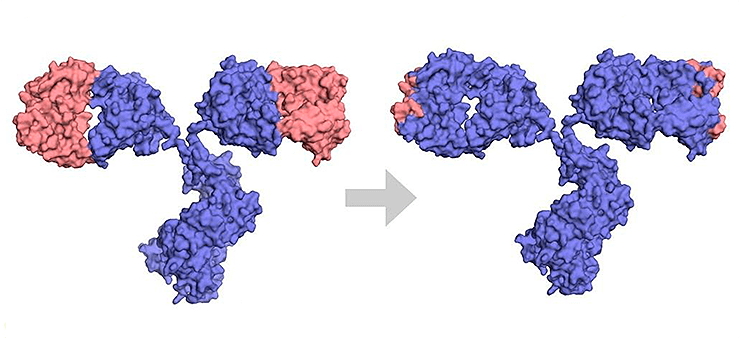
Рисунок 6. Разница между химерным и гуманизированным антителами. Синим показаны человеческие последовательности, оранжевым — мышиные.
Другой подход применили при разработке антитела даклизумаб против рецептора CD25 на Т-лимфоцитах, которое стало первым гуманизированным антителом на рынке (оно использовалось для лечения рассеянного склероза, но уступило конкурентам и было выведено с рынка). Даклизумаб также был получен включением мышиных CDR в человеческий каркас, но на этот раз человеческие участки антитела подобрали компьютерными методами так, чтобы максимизировать сходство с мышиным антителом, откуда взяты CDR. Кроме того, построили компьютерную модель мышиного антитела, и те аминокислотные остатки каркаса, которые контактировали с CDR, перенесли в каркас человеческого антитела, что позволило улучшить аффинность.
Хотя гуманизированные антитела включают повышенное количество человеческих последовательностей по сравнению с химерными, вероятность выработки антител против них остается — они называются HAHA (human anti-human antibody, человеческие анти-человеческие антитела).
HAMA-ответ может быть направлен на все антитело, HACA — против вариабельных областей, а HAHA-ответ еще более фокусный — только против CDR. Некоторые компании предприняли попытки заменить CDR-последовательности, чужеродные для человека, на человеческие. Таким образом удается повысить количество полностью человеческих последовательностей на 17–29% и получать с помощью гибридомной технологии почти полностью человеческие антитела.
Полностью человеческие антитела
Существует две группы технологий получения полностью человеческих антител. Одна из них подробно описана выше — это технологии дисплея, в первую очередь, фагового. Вторая — технология использования трансгенных животных, экспрессирующих человеческий репертуар антител. У метода трансгенных животных есть два важных преимущества перед дисплейными: антитела получаются быстрее и, поскольку отбор идет in vivo, а не in vitro, исключено образование антител с плохой растворимостью и другими проблемами в дальнейшей разработке.
Этим методом было получено одно из самых известных сейчас антител ниволумаб против мишени PD-1 на активированных T-лимфоцитах, которое вместе с другими антителами класса ингибиторов иммунологических чекпойнтов произвело революцию в лечении метастатических злокачественных заболеваний: меланомы, рака легкого, рака мочевого пузыря, дав впервые в истории надежду на полное выздоровление части пациентов. Ниволумаб был разработан по технологии компании Medarex, которая в 1993 году получила линию мышей, экспрессирующих человеческие IgM, IgG и Igκ и не экспрессирующих мышиные IgM и Igκ. Мыши оказались способными к производству полноценных В-клеток, в которых происходит V(D)J-рекомбинация человеческих трансгенных участков, а после введения антигенов — переключение класса тяжелой цепи и соматический мутагенез. Далее из этих мышей по обычной гибридомной технологии получают человеческие антитела.
Так, для получения ниволумаба трансгенных мышей иммунизировали рекомбинантным человеческим белком PD-1-Fc, состоящим из внеклеточного домена PD-1 и Fc-фрагмента IgG. Одновременно мышам ввели клетки яичников китайского хомячка CHO, экспрессирующие на поверхности PD-1. Спленоциты мышей слили с клетками миеломы SP2/0 и отобрали гибридомы, производящие антитела, реактивные против PD-1-Fc, с помощью ELISA. Связывание ниволумаба с CD4+ лимфоцитами определяли методом проточной цитофлуориметрии, а кинетику связывания с мишенью — методом плазмонного резонанса.
Несмотря на то, что у мышей технология Medarex порождает довольно ограниченный репертуар генов, кодирующих человеческие VH и VL, даже его хватает для генерации антител с высокой аффинностью, правда, не во всех случаях. Если антиген, к которому надо получить антитела, высокогомологичен с мышиным аналогом, сделать это не удастся. Здесь на помощь могут прийти трансгенные крысы, куры или дисплейные технологии. Также дисплейные технологии являются единственным выходом, если мишень токсична в концентрациях, необходимых для иммунизации животного, например, при разработке нейтрализующих антител против природных токсинов.
Изобретение множества методов разработки и гуманизации антител привело к тому, что граница между химерными, гуманизированными и человеческими антителами во многих случаях размыта. В связи с этим в 2017 году ВОЗ решила упразднить существовавшую ранее номенклатуру, по которой мышиные антитела имели суффикс -momab, химерные — -ximab, гуманзированные — -zumab, a полностью человеческие — -mumab.
Оптимизация константной части антитела
До сих пор речь шла в основном о том, как получить правильные последовательности CDR, которые определяют взаимодействие антитела с мишенью, его аффинность и отчасти иммуногенность. Теперь пришла пора поговорить о другой части антитела — Fc-фрагменте, структура которого определяет изотип антитела и такие важные его характеристики, как механизм действия (так называемые эффекторные функции), фармакокинетику и взаимодействие с остальными молекулами организма, помимо мишени.
Выбор подкласса IgG
У человеческого IgG четыре подкласса: IgG1, IgG2, IgG3 и IgG4 . Подклассы отличаются последовательностями Fc, и их особенности определяют профиль связывания с рецепторами на клетках иммунной системы — натуральных киллерах, нейтрофилах, макрофагах, а также связывание с белком системы комплемента C1q.
Механизмы действия антител
Клетки иммунной системы экспрессируют на поверхности семейство Fcγ-рецепторов, связывающихся с Fc-фрагментом антител подкласса IgG с разным сродством. Когда антитело связывается со своим антигеном, например, с рецептором на поверхности опухолевой клетки, конформация Fc-фрагмента немного меняется, и сродство к Fcγ-рецептору повышается. Клетка-эффектор с Fcγ-рецептором на поверхности присоединяется к антителу, и каскад событий внутри нее приводит к высвобождению цитотоксических гранул, которые убивают раковую клетку (рис. 7). Этот процесс носит название ADCC (antibody-dependent cytotoxicity — «антителозависимая цитотоксичность»). Наиболее активны в отношении ADCC натуральные киллеры (NK-клетки), несущие на поверхности рецептор FcγRIIIa. Помимо цитотоксических гранул, содержащих гранзим и перфорин, NK-клетки также выбрасывают цитокины IFNγ и TNF, создающие воспалительную среду и привлекающие другие клетки иммунной системы.
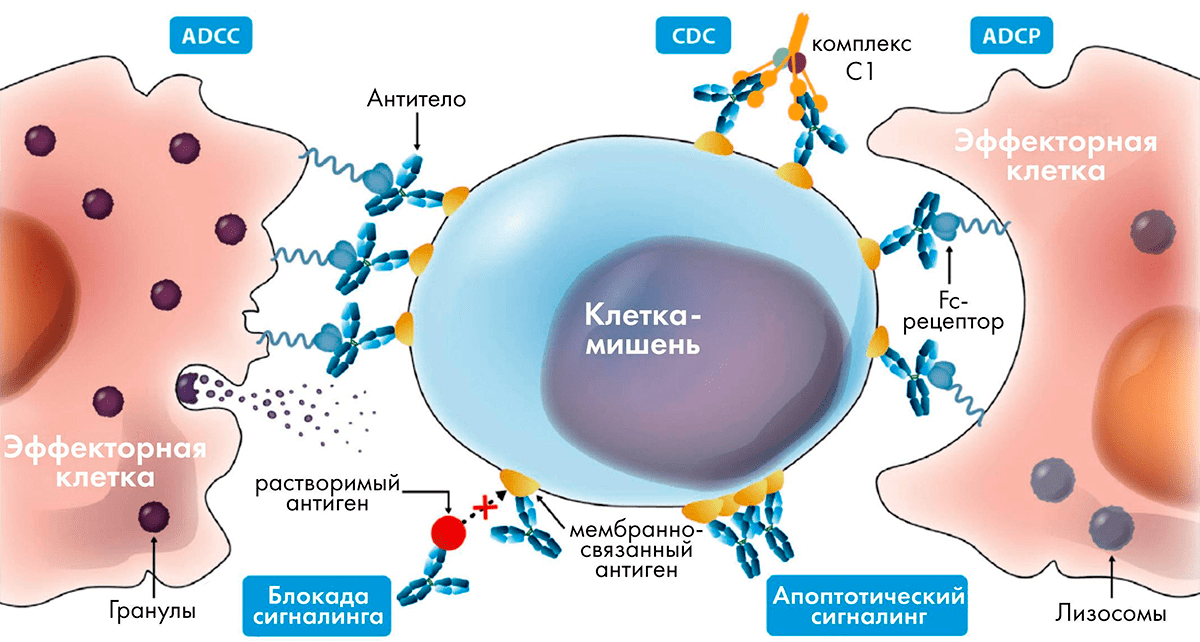
Рисунок 7. Различные механизмы действия антител
Показано, что ADCC играет важную роль в механизме действия многих антител против поверхностных мишеней, например, ритуксимаба (анти-CD20), трастузумаба (анти-HER2) и цетуксимаба (анти-EGFR). Наибольшее сродство к Fcγ-рецепторам имеет подкласс IgG1, поэтому, если необходимо, чтобы введение антитела приводило к уничтожению клеток, экспрессирующих мишень на поверхности, используют именно этот подкласс.
Сходный механизм — ADCP (antibody-dependent phagocytosis — антителозависимый фагоцитоз), при котором макрофаг после связывания Fc-фрагмента антитела с Fcγ-рецептором поглощает целиком и раковую клетку, и связавшееся с ней антитело.
Другой механизм уничтожения клеток, несущих мишень на поверхности, — CDC (complement-dependent cytotoxicity — комплемент-зависимая цитотоксичность). В этом случае с Fc-фрагментом связывается белок С1q системы комплемента, и запускается каскад событий, приводящий к образованию на поверхности клетки MAC-комплекса. MAC (membrane-associated complex) — это белки, которые формируют в мембране раковой клетки канал, что приводит к ее гибели.
Мутации в Fc-фрагменте IgG могут как увеличивать эффекторные функции — ADCC, ADCP и CDC, — так и снижать их. Так, оказалось, что ADCC антитела трастузумаб зависит от того, какой у пациента вариант рецептора FcγRIIIa. У пациентов с определенной мутацией в этом рецепторе антитело оказывается недостаточно эффективным. Введение мутаций в Fc-фрагмент трастузумаба повысило ADCC, что, возможно, позволит создать препарат, подходящий всем пациентам независимо от варианта FcγRIIIa.
В некоторых случаях необходимо только заблокировать мишень, например растворимые лиганды, при этом нет необходимости в гибели клеток. Тогда либо используют антитело другого подкласса — IgG2 или IgG4, которые почти не проявляют ADCC-активности, — либо вводят в Fc-фрагмент мутации, снижающие эффекторные функции. Так, для ниволумаба был выбран подкласс IgG4, потому что его мишень PD-1 экспрессируется в нормальных клетках иммунной системы, и было бы нежелательно их уничтожать. Вместо этого ниволумаб, не проявляя ADCC и CDC, блокирует взаимодействие PD-1 на Т-лимфоците с лигандом PD-L1 на поверхности раковой клетки и тем самым не дает раковой клетке снизить активность лимфоцита.
Гликозилирование
У молекул IgG в домене CH2 находится сайт N-гликозилирования, то есть присоединения остатков сахаров по атому азота (рис. 8). Модификация остатков сахаров, связанных с антителом, позволяет модулировать некоторые эффекторные функции.
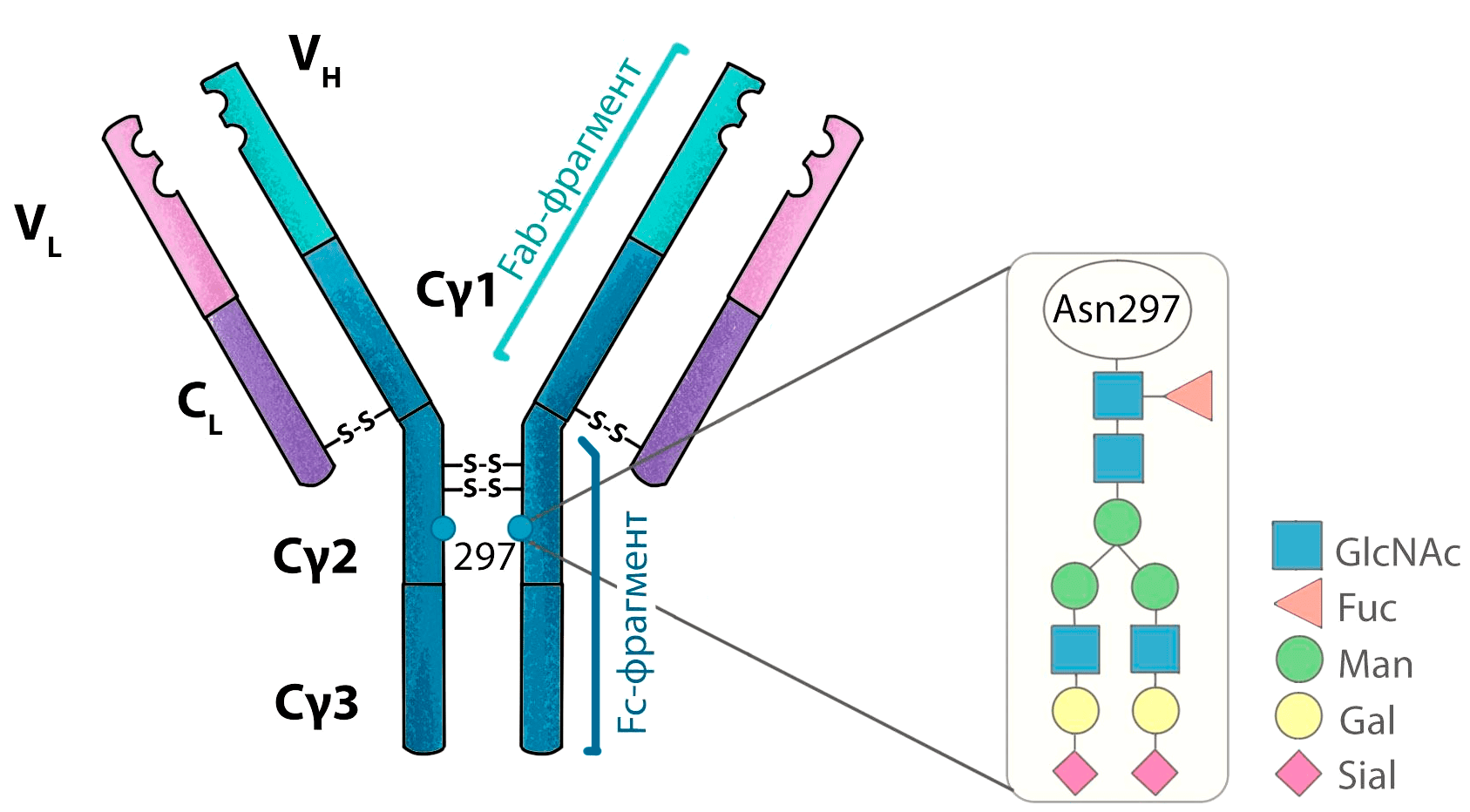
Рисунок 8. Структура гликана IgG. Гликозилирование антител происходит всегда в одном месте — по остатку аспарагина-297.
Гликозилирование абсолютно необходимо для взаимодействия антитела с Fcγ-рецепторами, поэтому удаление остатков сахаров приводит к тому, что CH2-домен изменяет свою конформацию, и антитело теряет все эффекторные свойства.
Модуляция состава сахаров, присоединенных к аспарагину-297, также позволяет увеличивать ADCC. К примеру, удаление остатка фукозы (дефукозилирование) приводит к росту ADCC. Чтобы добиться дефукозилирования, для производства антитела используют клеточную линию, в которой выключен фермент, отвечающий за добавление фукозы — фукозилтрансфераза.
Фармакокинетика
Специфические последовательности, расположенные в районе соединения доменов CH2 и CH3 молекулы IgG, отвечают за ее связывание с рецептором FcRn, которое в значительной мере определяет период полувыведения антитела. Этот удивительный рецептор экспрессируется на многих эндотелиальных и эпителиальных клетках и отвечает за транспорт антител через клеточные барьеры, а также за их возврат в кровоток. Связывание Fc-фрагмента антитела с FcRn в кровотоке при рН 7,4 незначительно, тогда как в кислой среде в эндосомах (pH < 6,5) связывание происходит. Клетки эндотелия постоянно захватывают белки плазмы в эндосомы, FcRn перехватывает антитела и возвращает их в циркуляцию. Если антитело не свяжется с FcRn, оно будет направлено на деградацию в лизосому (рис. 9).
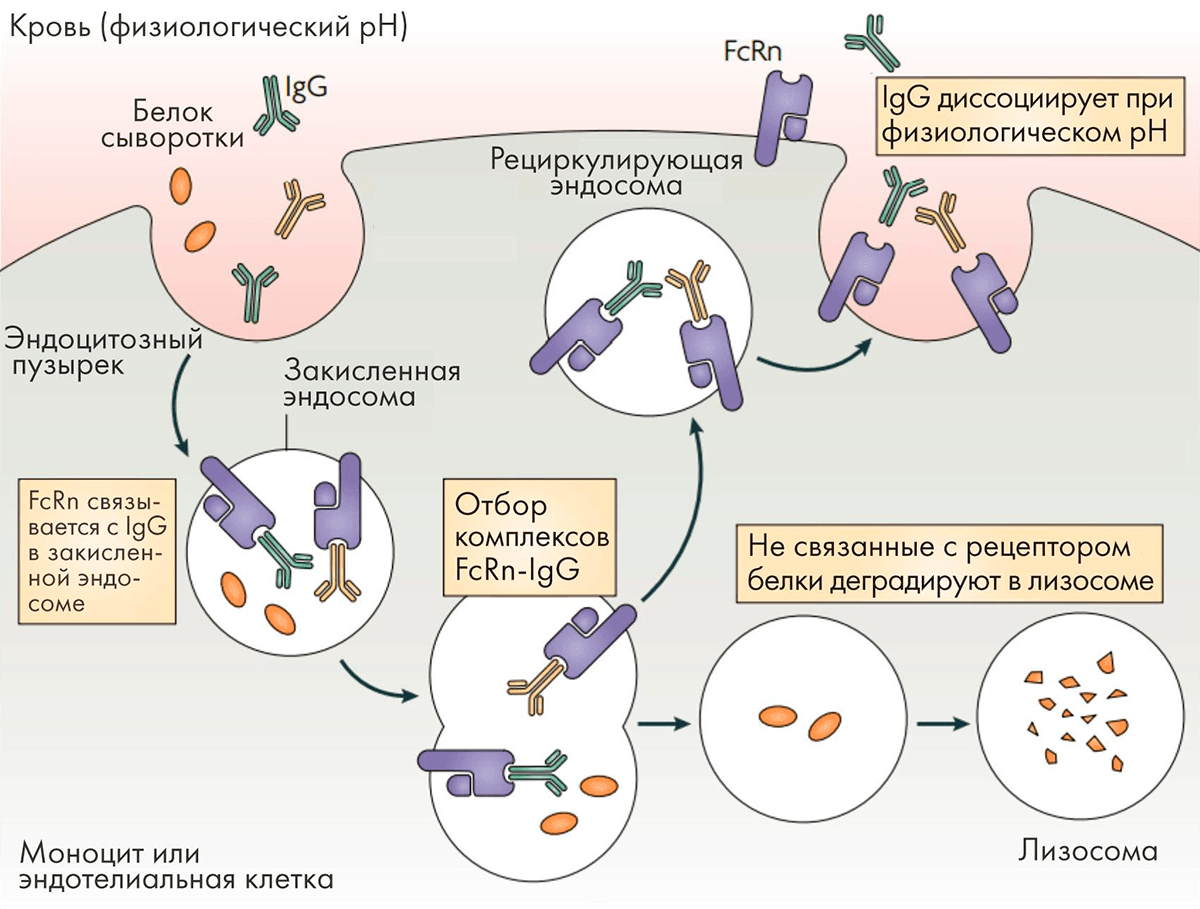
Рисунок 9. Взаимодействие антител с рецептором FcRn
Именно поэтому антитела подклассов IgG1, IgG2 и IgG4, обладающие высоким сродством к FcRn, имеют период полувыведения из организма 21 день, а антитела подкласса IgG3 с более низким сродством к нему — всего 5–7,5 дней. Были найдены мутации, усиливающие связывание IgG1 с FcRn, что позволило еще больше продлить период полувыведения.
Иногда, наоборот, нужно ускорить выведение антитела из организма, например, если оно потенциально токсично. В таких случаях вводят мутации, снижающие сродство к FcRn.
Методы изучения свойств антител
Плазмонный резонанс
Основной метод изучения кинетики и аффинности связывания антигена с антителом — поверхностный плазмонный резонанс (surface plasmon resonance, SPR). Метод основан на том, что на поверхности металлов при возбуждении источником света под определенным углом возникает плазмон — электромагнитная волна, распространяющаяся вдоль поверхности. Угол, при котором он возникает, зависит от свойств среды в непосредственной близи от поверхности, поэтому адсорбция веществ на поверхности металла приводит к изменению этого угла, и таким образом ее можно измерять (рис. 10).
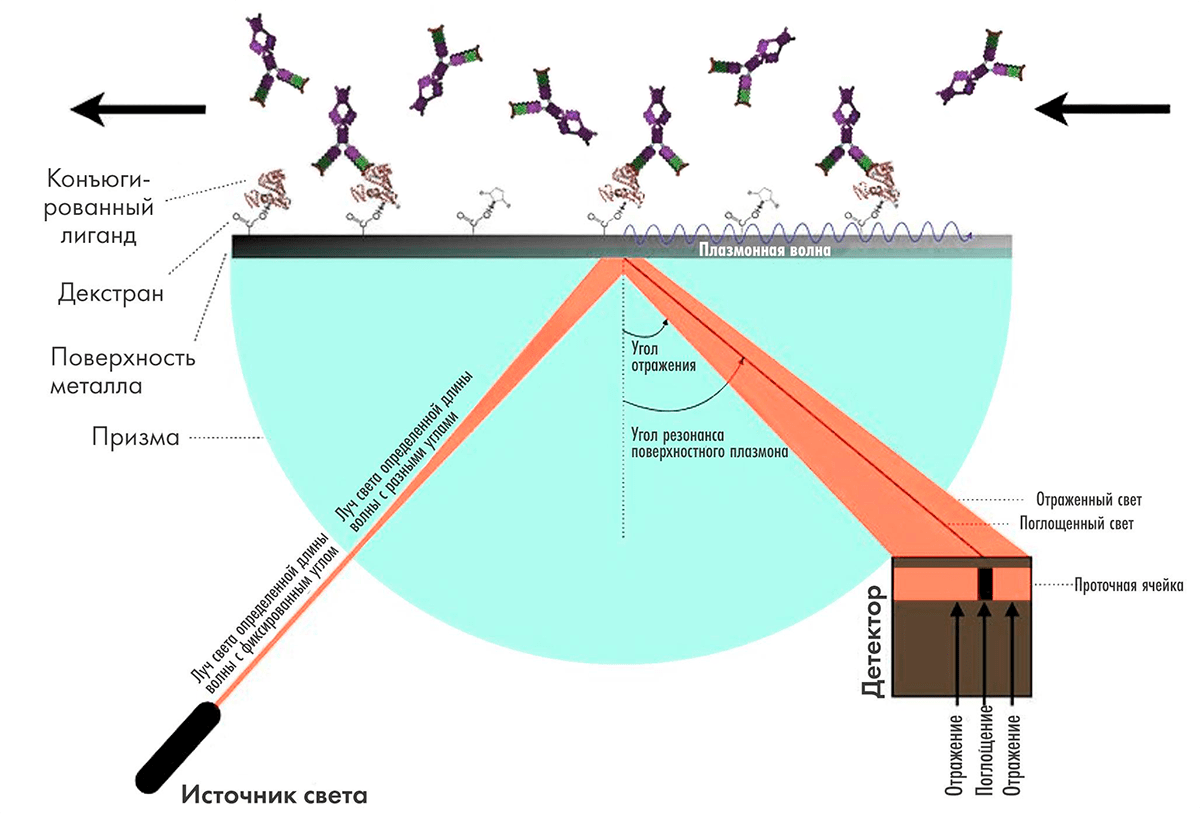
Рисунок 10. Схема работы прибора на принципе поверхностного плазмонного резонанса. Излучение от источника с длиной волны 760 нм отражается от металлической подложки, на противоположной поверхности которой закреплена одна из двух взаимодействующих молекул. Один из углов отражения является резонансным, и явление поверхностного плазмонного резонанса приводит к снижению интенсивности отраженного под этим углом света. При связывании антитела с конъюгированным лигандом меняется показатель преломления среды вблизи поверхности, резонансный угол отражения изменяется, что регистрируется фотоэлементом.
Наилучшие параметры метода, которые используются в наиболее популярных приборах для плазмонного резонанса Biacore, достигаются при применении в качестве поверхности золотых пластинок толщиной 50 нм. К слою золота ковалентно присоединяется декстрановая матрица, и молекула мишени иммобилизуется в декстране (разветвленном полимере глюкозы).
Антитело в буферном растворе пропускается над иммобилизованным лигандом, и связавшиеся антитела приводят к изменению резонансного сигнала. Метод SPR крайне чувствительный — предел обнаружения связывания антител составляет менее 10 пг/мм2.
Использование метода решает следующие основные задачи:
- обнаружение взаимодействия между молекулами;
- анализ кинетики взаимодействия;
- анализ аффинности;
- измерение концентраций;
- скрининг с целью поиска партнеров связывания.
Изучение плазмонного резонанса идеально подходит для скрининга, поскольку он не требует химических модификаций метками для измеряемых биологических молекул в растворе и хорошо автоматизируется.
Одним из ограничений метода является то, что вследствие иммобилизации области взаимодействия антигена с антителом могут экранироваться от антитела или возникнут пространственные затруднения для связывания. Это приведет к ложноотрицательным результатам. В таких случаях антиген модифицируют и используют те химические группы, которые находятся на удалении от сайта связывания с антителом или меняют антиген и антитело местами для вычитания стерического эффекта.
На измерения также может повлиять скорость переноса антитела из подвижной фазы (буфера) в неподвижную (декстрановую матрицу), поэтому для подтверждения результата рекомендуется пользоваться независимым методом, например, KinExA, в котором измеряется взаимодействие антитела не с иммобилизованным антигеном, а со свободным.
Вторая проблема заключается в том, что свойства антитела и антигена в растворе и на поверхности отличаются от соответствующих свойств в организме, однако это фундаментальное затруднение характерно для всех аналитических методов in vitro.
Проточная цитофлуориметрия
Другой метод, который рутинно используют при разработке антител — FACS (fluorescence-activated cell-sorting — сортировка клеток с активацией флуоресценции или, по-другому, проточная цитофлуориметрия). В случае антител с помощью FACS можно определить аффинность связывания с поверхностным рецептором клетки прямо на клеточной поверхности, не выделяя рецептор. Это очень полезная особенность, так как поверхностные рецепторы являются мембранными белками, и манипуляции с ними in vitro сопряжены со множеством трудностей: для них требуется создавать особые условия, иначе они не сохраняют свою конформацию и агрегируют.
Для определения аффинности постоянное количество клеток, несущих рецептор, титруется возрастающей концентрацией антитела, при этом каждый раз устанавливается равновесие. Антитела, связавшиеся с клетками, детектируют с помощью других антител, меченых флуоресцентной меткой.
В связи с хорошей автоматизируемостью и высокой производительностью метода он хорошо подходит для скрининга библиотек клонов антител. Один и тот же клон проверяют на связывание с клетками, экспрессирующими и не экспрессирующими рецептор. Если отношение связывания больше определенного порога, клон отбирают для дальнейшей работы.
Методы in silico
После того, как методом дисплея получены фрагменты антител с высокой аффинностью к мишени, важно провести картирование эпитопов, то есть определение того, с каким участком мишени связываются антитела-кандидаты. Кандидаты, связывающиеся с разными участками мишени, лучше подходят для дальнейшего тестирования, так как в случае неудачи одного из кандидатов больше шансов, что другой кандидат будет успешен.
Экспериментальное картирование, например, с помощью мутагенеза или рентгено-структурного анализа — долгий и трудоемкий процесс, который не подходит для анализа кандидатов на начальном этапе. Поэтому прибегают к картированию эпитопов in silico — то есть компьютерному расчету эпитопов исходя из аминокислотной последовательности мишени, антиген-связывающего фрагмента антитела и сведений, содержащихся в базах данных эпитопов. Алгоритмы на основе машинного обучения и больших баз данных белковых последовательностей и трехмерных моделей способны предсказывать как линейные, так и конформационные эпитопы. Точность таких предсказаний не стопроцентна, но для грубого отбора фрагментов-кандидатов она вполне достаточна.
Методы in silico применяют и на других стадиях разработки антител. Так, для поиска аминокислотных остатков, предположительно обусловливающих агрегацию антител, используют метод SAP (spatial aggregation propensity — пространственную склонность к агрегации). По этой технологии каждой аминокислоте в антителе присваивается величина SAP в зависимости от ее гидрофобности, степени доступности на поверхности, вкладов гидрофобных взаимодействий с другими аминокислотами в пределах заданного радиуса. Затем для антитела строится цветовая карта, из которой сразу видно, какие области молекулы отвечают за агрегацию. Экспериментально показано, что замена аминокислот в таких участках на более гидрофильные позволяет снизить агрегацию белка и тем самым улучшить его фармакологические свойства.
Для разработки стратегии гуманизации антитела также используют методы in silico. При вставке мышиной вариабельной последовательности в человеческий каркас аффинность антитела к антигену может быть снижена. В этом случае применяют «обратное» введение мутаций, отвечающих за более сильное связывание с антигеном. Такие мутации, в частности, можно выбирать путем статистического анализа последовательностей известных комплексов антитело—антиген.
Методы молекулярного моделирования с использованием расчета свободной энергии связывания антител с антигенами иногда используют на этапе созревания аффинности для поиска мутаций, обеспечивающих более высокую аффинность. Эти методы предполагают применение энергетических функций, отражающих взаимодействия атомов антигена и антитела между собой. Такие функции могут быть или основаны на физических параметрах взаимодействия атомов, или рассчитаны исходя из статистических данных — как правило, извлеченных из трехмерных структур комплексов антиген—антитело. Однако из-за сложности объекта моделирования и неизбежности многих допущений успехи в этой области пока не так велики, как хотелось бы.
Производство антител
Итак, мы получили генетическую конструкцию, кодирующую антитело с нужными нам свойствами, ввели ее в клетку-продуцент, получили в лаборатории нужные антитела и охарактеризовали их. Что же дальше? Теперь необходимо масштабировать производство и очистку антитела, чтобы получить его в достаточных количествах для последующих исследований, а потом, если все пойдет хорошо, и для продажи. Причем если в случае лабораторных процессов исследователи сами следят за их качеством, то как только лекарственный препарат доходит до стадии исследования токсичности на животных, он попадает под действие законодательства: регуляторные органы (FDA в США, Минздрав в РФ, EMA — Европейское медицинское агентство — в Евросоюзе и т. д.) принимают только такие исследования, которые соответствуют строгому набору правил. В частности, препарат должен быть произведен в соответствии с правилами GMP — Good manufacturing practice («Надлежащей производственной практики»). GMP регламентирует все аспекты производства, начиная от требований к устройству помещений и заканчивая контролем качества продукции. Регулятор имеет право в любой момент проверить соблюдение правил GMP производителем лекарства.
При производстве антител разработчики должны учитывать следующие критерии:
- качество получаемого продукта;
- себестоимость производства;
- время и сложность постановки процесса.
Получение клона-продуцента
Для получения стабильного клона-продуцента антител разработчикам нужно добиться правильного встраивания вектора в геном клетки, модифицировать, если необходимо, аппарат трансляции и посттрансляционных модификаций, добиться высокой жизнеспособности продуцента. В клетках CHO с этой целью снижали экспрессию про-апоптотических факторов и повышали экспрессию анти-апоптотических (например Bcl-2). Трансляцию белка увеличивали, сверхэкспрессируя шапероны, чтобы защитить клетку от апоптоза под воздействием стресса и обеспечить более правильное сворачивание антител. Кроме того, в геном клетки-продуцента вносят модификации, обеспечивающие желаемый профиль гликозилирования.
После создания клеточной линии начинается подбор и оптимизация процесса культивирования, сначала в 96-луночных планшетах, затем в колбах и настольных биореакторах объемом 1–5 л. В результате скрининга сотен и тысяч клонов выбирают клон-продуцент с наилучшими характеристиками и переходят к пилотному культивированию в реакторах объемом 10–25 л, а затем масштабируют процесс до бóльших размеров (рис. 11).
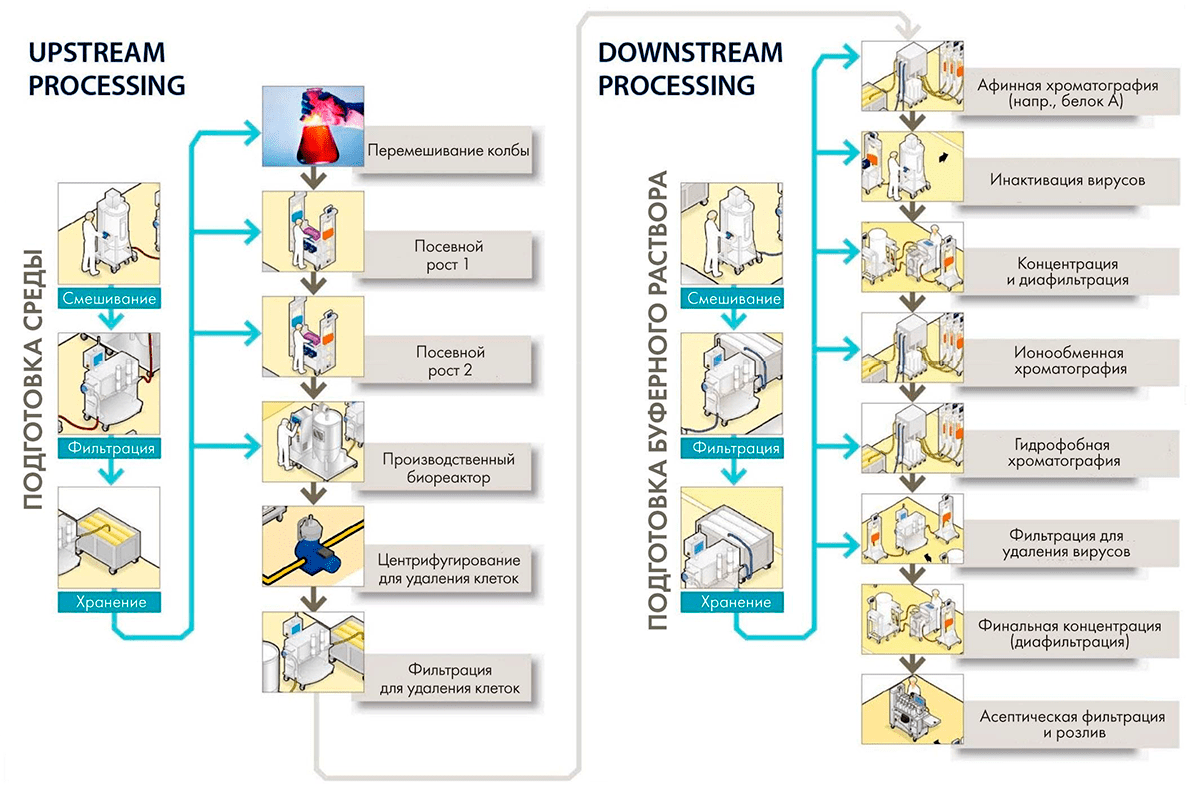
Рисунок 11. Производство антител и других рекомбинантных белков делится на две большие стадии: upstream process (USP), включающий все стадии от размораживания пробирки с клоном-продуцентом до сбора среды культивирования, и downstream process (DSP) — процесс выделения и очистки продукта.
Культивирование
Обычно клетки, производящие антитела, культивируют в биореакторах-ферментёрах объемом от 100 до 25 000 л (рис. 12). Традиционным способом культивирования являются процессы batch и fed-batch. В первом случае все компоненты среды и клон-продуцент загружают в реактор на несколько дней, после чего собирают культуральную среду для выделения белка. Во втором случае в среду периодически добавляют питательные вещества.

Рисунок 12. Внешний вид биореактора
Важнейшим фактором обеспечения высоких выходов белка является культуральная среда. Причем прогресс в этой области продолжается, и если в 1990-х годах выходы составляли 0,1 г/л, в 2007 — до 5 г/л, то сейчас удается достигать 10 и более г/л. Такие успехи стали возможны благодаря переходу от сред, содержащих сыворотку животных к бессывороточным: сначала использовались гидролизаты белков, а сейчас — полностью химические среды с четко определенным составом. Дело в том, что вариабельность состава ухудшает параметры культивирования. Также оказалось плодотворным изменение состава среды в зависимости от стадии культивирования: применяются разные среды в фазе роста и в фазе накопления продукта.
Для повышения продуктивности применяют также тонкую настройку параметров среды с обратной связью: температуры, рН, солевого состава, уровней O2 и CO2, что позволяет добиться снижения концентрации токсичных побочных продуктов (молочной кислоты, аммиака) и более плавного расхода питательных веществ.
В настоящее время наблюдается тенденция к переходу на непрерывные процессы: на так называемое перфузионное культивирование, при котором свежая среда непрерывно добавляется к клеточной культуре, а продукт удаляется из нее. Такой подход лучше автоматизируется, дает продукт более высокого качества и с увеличенными выходами. Также разработчики переходят от стальных биореакторов к одноразовым контейнерам, так как у последних есть ряд преимуществ:
- они поставляются чистыми и стерильными, их не нужно готовить к работе;
- по этой же причине проще валидировать процесс производства;
- они обладают большей гибкостью в случае необходимости изменения параметров процесса;
- их не нужно долго вводить в эксплуатацию и очищать в случае контаминации.
Выделение и очистка антител
Антитела секретируются клетками-продуцентами в окружающую среду, и для получения чистого препарата требуется многоступенчатый процесс отделения антитела от всех ненужных и потенциально опасных примесей (рис. 11). Основными стадиями здесь являются различные виды хроматографии, диафильтрация и инактивация вирусов. Одна из особенностей очистки антител по сравнению с другими белками — использование хроматографии с белком А. Это белок клеточной стенки золотистого стафилококка, который сильно и селективно связывается с антителами при нормальном рН и слабо — при пониженном. Благодаря высоким выходам и чистоте получающегося продукта это предпочтительный метод на первом этапе очистки антител.
Важнейшим требованием и на стадии производства, и на стадии очистки антитела является постоянство состава продукта. Поскольку антитело — сложный объект и производится в биологической системе (в клетках), молекулы получаются не одинаковые, а с вариациями (рис. 13). Цель разработки процесса производства и очистки — добиться того, чтобы эти вариации не влияли на фармакологические характеристики продукта, его стабильность, и не слишком менялись от одной партии продукта к другой. Для этого проводится валидация процесса производства — экспериментальное подтверждение того, что процесс обеспечивает получение продукта с надлежащими характеристиками.
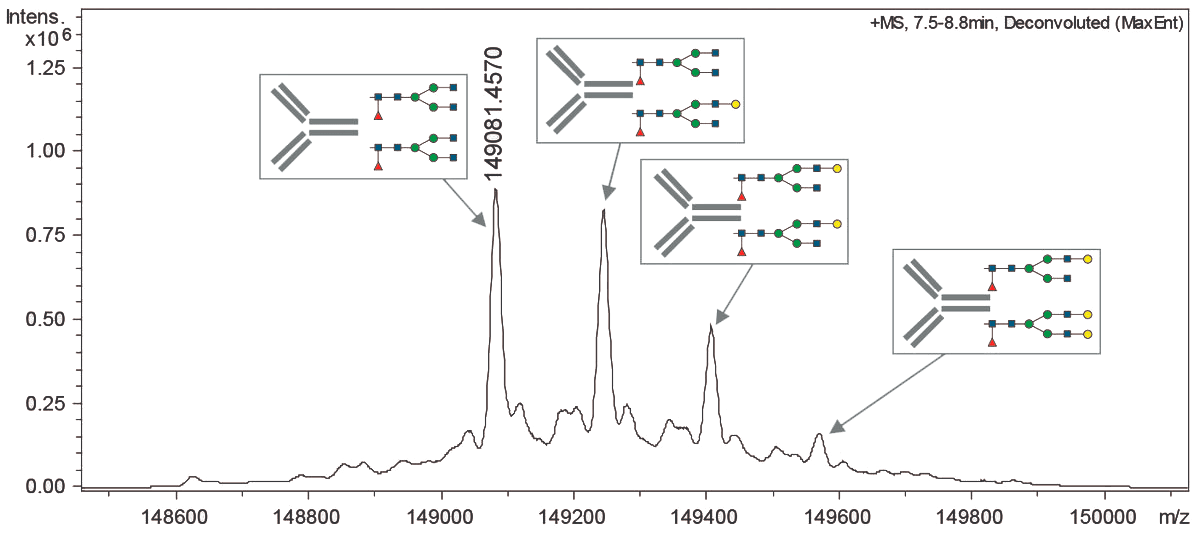
Рисунок 13. Профиль гликозилирования определяют методом масс-спектрометрии. На этом спектре видна типичная картина гетерогенности препарата антитела.
На некоторых промежуточных и на финальном этапе состав продукта контролируют аналитическими методами. Обязательно следят за:
- бактериальными эндотоксинами;
- белком А;
- наличием вирусов;
- белками и ДНК продуцента, остатками культуры (антибиотиками, сывороткой и пр.), остатками даунстрим-процессинга (ферментами; реагентами — цианогенбромидом, гуанидином и пр.; неорганическими солями, растворителями, носителями, лигандами и пр.).
- физико-химическими свойствами (углеводным составом, дисульфидными мостиками, вариабельностью N- и С-концевых остатков, четвертичной структурой);
- наличием изоформ, коэффициентом экстинкции, электрофоретическим, хроматографическим, спектроскопическим профилями, мультимерами и агрегатами, гетерогенностью заряда молекулы.
Часть этих показателей входит в спецификацию на субстанцию, из которой затем получают лекарственный препарат.
Методы производства антител все время совершенствуются: за 40 с лишним лет, прошедших с момента вывода первого антитела на рынок, изменилось многое — антитела научились гуманизировать, разработали способы их искусственного получения, усовершенствовали методы изучения аффинности и других свойств, достигли огромного прогресса в производстве и очистке, который позволил снизить себестоимость антител ниже $500 за 1 г. Несмотря на все эти успехи, существует потребность в более быстрой и точной разработке антител против заданной мишени. В идеале было бы как можно больше операций проводить in silico, чтобы не тратить время и деньги на эксперименты, но пока у нас недостаточно знаний и мощностей для моделирования, например, фармакокинетики и фармакодинамики антител. Тем не менее описанные в этой статье технологии уже позволили создать десятки лекарственных препаратов, которые помогают миллионам людей.
Источник: БИОМОЛЕКУЛА
