
Сегодня диагностика рака требует анализа пробы ткани — биопсии. И несмотря на то, что эту процедуру зачастую уже можно выполнить малоинвазивно, она малоприятна и может дать ложноотрицательный результат из-за того, что будет проведена неаккуратно и «не попадет» в опухоль. Кроме того, в случае отрицательного результата (когда худшие подозрения не подтвердились) биопсия увеличивает риск развития рака в будущем, а для некоторых пациентов забор материала просто невозможен. Всех этих и многих других недостатков позволяет избежать так называемая «жидкая биопсия» — анализ циркулирующих в крови молекул и опухолевых клеток. Она позволяет определять диагноз, прогноз и курс лечения, правда, точность технологии пока оставляет желать лучшего. Тем не менее уже известны циркулирующие эпигенетические биомаркеры, позволяющие диагностировать рак на ранней стадии (к примеру, анализ метилирования гена SEPT9). Недавнее исследование российских ученых, подтвержденное зарубежными коллегами, позволяет в ходе «жидкой биопсии» идентифицировать целый пласт источников циркулирующих биомаркеров. Спектр новых возможностей метода еще предстоит изучить, а основа для
Геном человека состоит из 46 хромосом — длинных линейных отрезков ДНК, плотно и иерархически (в несколько этапов) упакованных с помощью специальных белков в компактные образования. Минимальная единица этой иерархической упаковки — нуклеосома. Нуклеосома — это комплекс из восьми молекул гистонов (класс ядерных белков), похожий на катушку, вокруг которой приблизительно в 1,7 оборота намотано 150 нуклеотидных пар (п.н.) ДНК (рис. 1). Таким образом, ДНК в наших клетках напоминает бусы из «катушек»-нуклеосом.
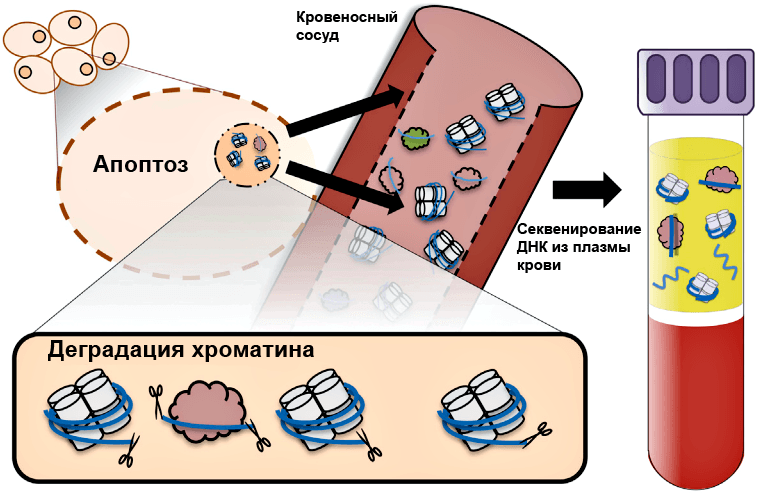
Рисунок 1. Схематическое изображение источника ДНК в плазме крови. В ядре ДНК вместе с гистонами образует хроматин — плотно упакованный комплекс (гистоны изображены в виде белых цилиндров, ДНК — в виде голубой нити). Плотность упаковки зависит от необходимости доступа к ДНК: «молчащие» гены плотно, зачастую хаотично упакованы, в то время как для активно экспрессируемых генов характерна упорядоченная эпигенетическая структура. В процессе апоптоза деградация ДНК (ножницы — эндонуклеазы) осуществляется по свободным участкам, не связанным с белками — гистонами или транскрипционными факторами (облачкá). Деградированная ДНК с помощью везикулярных телец высвобождается в кровь, где впоследствии может быть обнаружена с помощью гель-электрофореза.
Поскольку считывать генетическую информацию можно только с ненамотанной ДНК, подобная укладка оказывает огромное влияние на уровень экспрессии генов. Значение имеет и тип гистонов, из которых состоят нуклеосомы, и претерпеваемые ими модификации, и место расположения на ДНК. В основной массе эти белки хаотично расположены по всему геному, постоянно меняя свое место связывания, однако есть участки ДНК, требующие строго определенного позиционирования гистонов.
Наиболее изучены в этом плане места начала транскрипции (transcription start sites, TSS). На клеточных линиях дрожжей и T-лимфоцитов человека ранее было показано, что у активно экспрессируемых генов нуклеосома жестко расположена в районе 100 п.н. в направлении 5′→3′ от TSS и нет нуклеосомы в районе 50 п.н. в обратном направлении (рис. 2), в то время как у «молчащих» генов нуклеосомы расположены в области TSS хаотически. Такой паттерн укладки нуклеосом, скорее всего, связан с необходимостью организации транскрипционного комплекса: гистоны конкурируют с транскрипционными факторами, ДНК-полимеразой и прочими белками за место посадки. Таким образом, расположение нуклеосом в гене указывает на то, как активно он экспрессируется в данной клетке.
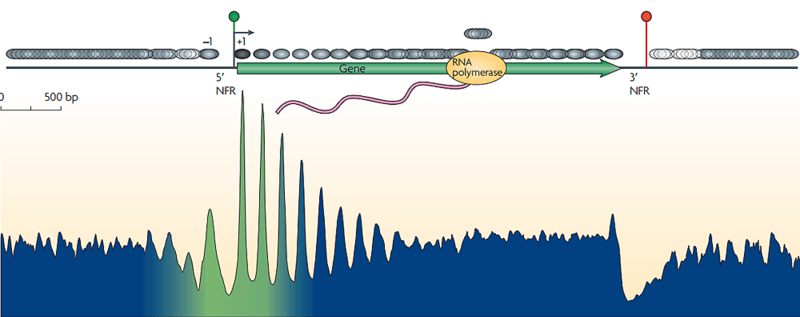
Рисунок 2. Консенсусное распределение нуклеосом среди всех генов дрожжей, выровненных по началу гена.
Остается вопрос — как узнать о расположении нуклеосом без непосредственного доступа к клетке организма? Ответ дает апоптоз (запрограммированная клеточная смерть): во время апоптоза ДНК претерпевает конденсацию и ферментативную деградацию эндонуклеазами. Поскольку гистоны, как уже отмечалось выше, конкурируют с прочими белками за связывание с ДНК, то расщепляющие ее ферменты имеют доступ в основном только к участкам ДНК между нуклеосомами — так называемым линкерам, — по ним ДНК и разрезается (рис. 1). Впоследствии с помощью везикулярных телец деградированная ДНК высвобождается в кровь, где она либо связывается с мембранными белками форменных элементов в сыворотке, либо свободно «плавает» в плазме. Таким образом, циркулирующая в крови ДНК (цирДНК) представлена квантующимися размерами: превалируют мононуклеосомы, потом ди-, тринуклеосомы и так далее, формируя при электрофорезе в агарозном геле «апоптотическую лестницу» — отдельные полосы, соответствующие различным по молекулярной массе фрагментам.
Далее, если мы проанализируем последовательности коротких фрагментов ДНК в крови и определим, откуда они пришли из генома (картируем по референсному геному человека), то сможем определить ту самую картину нуклеосомной укладки в ткани — источнике ДНК (паттерн нуклеосомной укладки). А поскольку знание паттерна нуклеосомной укладки позволяет судить об уровне экспрессии гена, можно, к примеру, обнаружить признаки аномальной экспрессии генов опухолевых супрессоров или онкогенов, что указывает на развитие опухолевого процесса.
Идея проста на словах. В реальности же помимо нуклеосомной укладки на характер деградации ДНК влияет множество факторов, а сама ДНК в крови человека представляет собой смесь фрагментов из множества тканей-источников: она собирается в кровь со всего организма, а в разных тканях разные гены экспрессируются по-разному, поэтому картины накладываются друг на друга. Таким образом, до сих пор не удавалось продемонстрировать возможность реализации этой идеи.
В течение последнего десятилетия активно велись исследования методик анализа паттерна фрагментации ДНК на клеточных линиях — дрожжей и клеток крови человека. Впервые к анализу непосредственно цирДНК подобрался коллектив российских ученых из Медико-Генетического Научного Центра, Университета Джорджа Мэйсона и компании «Атлас-Онкодиагностика»: они использовали данные высокопроизводительного секвенирования (NGS) экзома цирДНК, выделенной из плазмы двух пациентов со злокачественными новообразованиями. То, что казалось проблемой, удалось обернуть себе на пользу: поскольку цирДНК представляет собой смесь ДНК из всех тканей организма, если взять гены, активно экспрессируемые во всех тканях (так называемые гены домашнего хозяйства), и посмотреть на их паттерн нуклеосомной укладки по сравнению с тканеспецифичными генами, то мы должны ожидать, что первые будут демонстрировать картину, характерную для активно экспрессируемых генов, а вторые — для «молчащих», так как в большинстве тканей они активно не работают. И эту разницу удалось увидеть: отношение высоты +1 пика (рис. 2) к последующему минимуму было значительно больше для генов домашнего хозяйства (рис. 3). Так удалось подтвердить новую «биомаркерную концепцию» — по ДНК из плазмы крови можно воспроизвести картину нуклеосомной укладки в тканях и судить об особенностях эпигенетической регуляции.
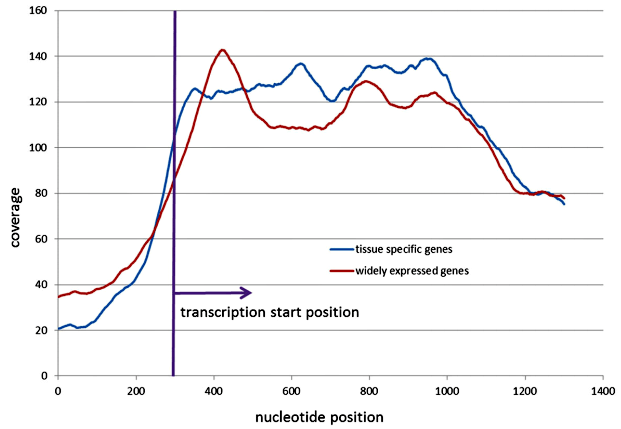
Рисунок 3. Консенсусное распределение покрытия в области TSS генов домашнего хозяйства (widely expressed genes) и тканеспецифичных генов (tissue specific genes) для циркулирующей ДНК и геномного контроля. Характер кривой генов домашнего хозяйства соответствует характерному положению нуклеосом в активно экспрессируемых генах, в то время как кривая тканеспецифичных указывает на отсутствие устойчивого нуклеосомного паттерна, что характерно для «молчащих» генов.
Спустя месяц после публикации российских исследователей, в Cell вышла статья с новой информацией в поддержку этой концепции: получив большой объем данных секвенирования плазмы крови больных и здоровых, удалось идентифицировать даже ткань, проявляющую аномальную экспрессию. Для 44 образцов плазмы крови пациентов с диагностированным раком IV стадии (твердые опухоли различных органов и гистологии) провели полногеномное секвенирование. Для каждой позиции генома считали сигнал L-WPS — количество фрагментов, полностью перекрывающихся с окном в 120 п.н., центрированным по позиции. Далее полученный сигнал обрабатывали с помощью быстрого преобразования Фурье. Среднюю интенсивность сигнала в окне частот 193–199 п.н. для преобразования первых 10 т.п.н. гена принимали за меру его экспрессии. Затем вычисляли корреляцию посчитанных таким образом профилей экспрессии и заранее известных профилей экспрессии клеточных линий. Результаты приведены на рисунке 4.
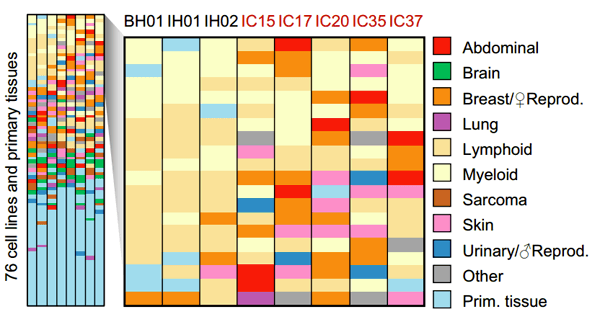
Рисунок 4. Корреляция 76 известных профилей экспрессии различных клеточных линий с предсказанными по паттерну фрагментации циркулирующей ДНК. Каждый столбец соответствует отдельному образцу плазмы крови, в столбцах — профили экспрессии различных клеточных линий (перечислены справа), упорядоченные по убыванию корреляции. BH01, IH01, IH02 — здоровые люди. В их случае особенно сильна корреляция с профилями экспрессии клеточных линий лимфоидного происхождения, что очевидно — кровь вносит наибольший вклад в состав цирДНК. IC15, IC17, IC20, IC35 и IC37 — пациенты с диагностированным раком. Для них среди первых 10 тканей с наивысшей корреляцией уже встречаются клеточные линии соответствующей патологии: IC17 (рак печени) — HepG2 (клеточные линии HCC), IC35 (рак груди) — MCF7 (клеточная линия распространенной аденокарциномы груди) и т.д. Таким образом удается не только уловить следы опухоли в крови, но и предположить, какого она происхождения.
Благодаря результатам российских ученых, подтвержденным в работе зарубежных коллег, появляются новые возможности неинвазивной диагностики онкологических заболеваний и открывается новый пласт фундаментальных исследований, которые, возможно, смогут пролить свет не только на онкологические, но и на другие патологические процессы.
Источник: БИОМОЛЕКУЛА
